科研进展 JCTC | 利用混合量子-经典计算进行大规模分子几何结构优化
近日,北京大学袁骁团队、电子科大王晓霆团队等联合研究团队,提出了一种全新的“量子-经典协同优化框架”,成功实现了乙醇酸(Glycolic Acid)等复杂分子的几何结构优化,首次实现大尺度药物分子的几何优化的量子算法。该成果以 “Large-Scale Efficient Molecule Geometry Optimization with Hybrid Quantum-Classical Computing” 为题,发表于中科院1区、理论计算化学领域的顶级期刊 《Journal of Chemical Theory and Computation》。
研究背景
分子构型的高精度预测是量子化学的灵魂,它直接关联着物性机理研究与创新药研发的底层逻辑。 尽管高精度电子结构理论(如 CCSD 或 MRCI)在微观世界表现卓越,但面对工业级大分子体系时,其算力需求会陷入“指数级增长”的泥淖,使实际计算变得难以为继。尽管量子计算有望重塑计算范式,但在当前的 NISQ 时代,技术落地仍面临双重困境。其一,硬件规模的局限导致有效比特数难以覆盖复杂分子轨道,制约了模拟上限;其二,算法架构的冗余,目前主流的几何优化仍高度依赖“坐标变换-能量重算”的嵌套迭代,这种步进式逻辑不仅由于计算频次过高而放大了噪声干扰,更导致收敛效率低下。因此,探索能够跨越硬件瓶颈与算法冗余的新路径,已成为当前的科研攻关核心。
方法概述
北京大学袁骁团队、电子科大王晓霆团队等团队联合攻关,构建了“DMET-VQE 融合计算框架”。 这一框架的核心竞争力在于对计算逻辑的系统性重塑。团队首先利用 DMET 策略对大分子进行“碎片化”处理,在保留核心电子相干性的前提下,将复杂的全局模拟转化为多个低位比特需求的局部任务。而在优化路径上,该研究摒弃了传统的“外循环优化”模式。 团队创新性地开发了协同优化(Co-optimization)技术,让核坐标与量子线路参数在同一个闭环内实时更新。这种设计告别了“先算能量、再调结构”的冗余模式,极大地削减了对量子硬件的调用频率。该研究不仅证明了量子算法在处理工业级分子体系时的潜力,更为跨越 NISQ 时代的计算障碍指明了方向。
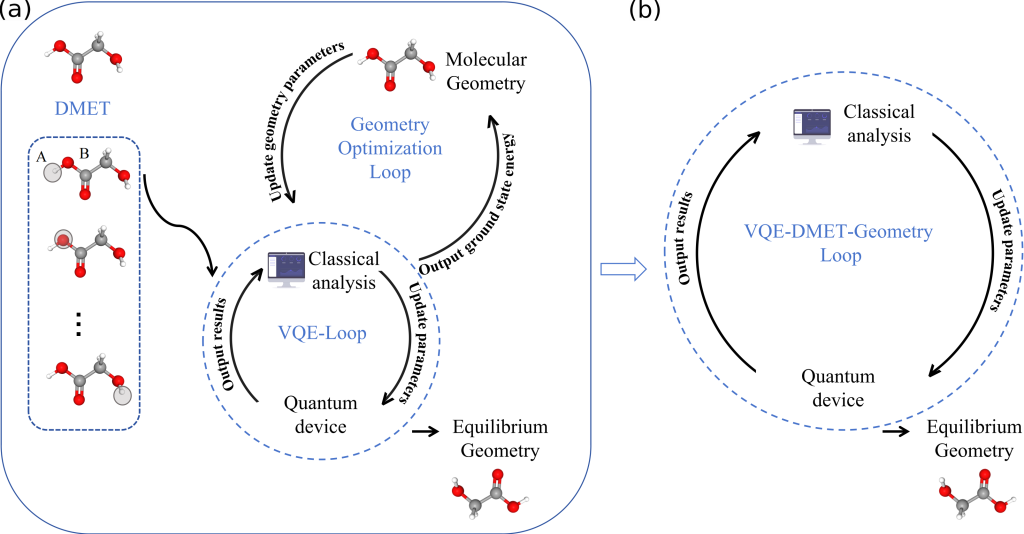
研究成果
该协同优化框架在多个具有代表性的分子体系中展现了卓越的性能与巨大的应用潜力。在首先完成了对 H4 和过氧化氢等基准分子的有效性验证后,研究团队成功将该算法应用于更具挑战性的生物相关分子——乙醇酸的几何构型优化中 。
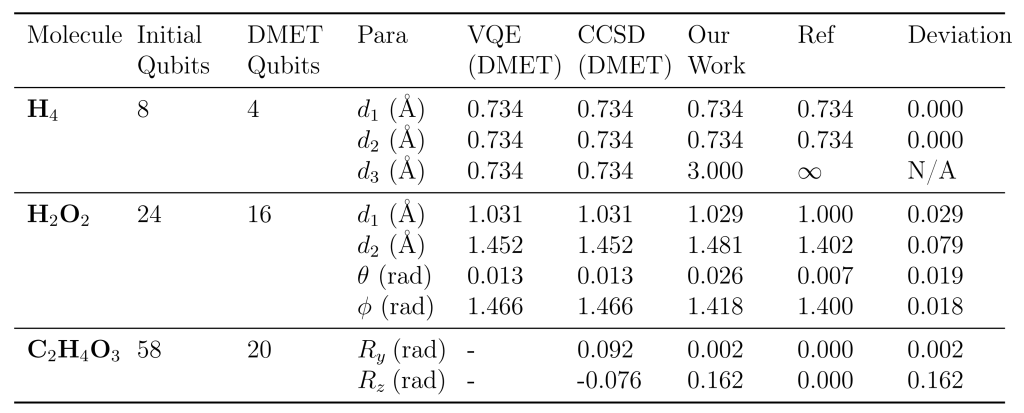
实验数据表明,对于乙醇酸这样复杂的体系,传统全量模拟所需的量子比特数高达 58 个,而利用该团队研发的 DMET 嵌入策略,所需比特数被惊人地压缩至 20 个,成功使其落入现有量子硬件的可运行范围内 。在精度方面,协同优化算法表现优异,其计算得到的键长、键角及关键的羟基旋转角度与经典的高精度耦合簇理论(CCSD)计算结果高度吻合,偏差微乎其微 。轨迹分析进一步显示,相比于传统的嵌套优化方案,协同优化策略能够沿着能量曲面更平滑、更快速地收敛至平衡态,显著减少了迭代步数 。这也是目前已知首次利用量子算法成功优化如此规模分子的几何结构,标志着量子计算辅助复杂药物分子与催化剂设计方面取得了里程碑式的进展。
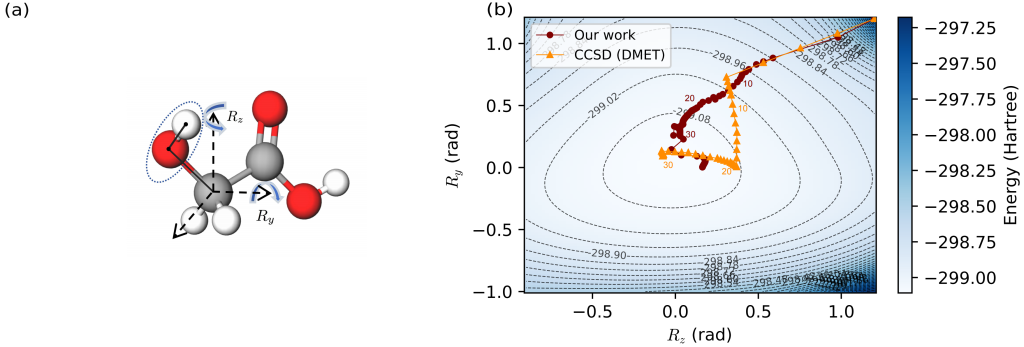
研究意义
这项研究成果不仅是算法层面的技术革新,更是量子计算迈向实用化的重要工作。通过成功突破传统量子模拟对量子比特数量和计算时间的双重限制,研究团队证明了在当前的小规模量子设备上处理具有真实化学意义的复杂分子是完全可行的。协同优化框架的提出,为利用量子优势进行复杂催化剂、药物分子及光伏材料的“计算机辅助设计”(in silico design)建立了一条切实可行的路径,极大地拓展了量子模拟在制药与化工行业的应用。此外,这种基于 DMET 的协同优化策略具有极强的通用性与扩展性,未来可进一步结合先进的误差缓解技术与高性能量子硬件,深入探索强关联电子体系等传统经典方法束手无策的领域。
Yajie Hao, Qiming Ding,∗ Xiaoting Wang,∗ and Xiao Yuan∗. “Large-Scale Efficient Molecule Geometry Optimization with Hybrid Quantum–Classical Computing.” J. Chem. Theory Comput. 2026, 22, 2, 859–868
doi:https://doi.org/10.1021/acs.jctc.5c01435